declump those buggers


|
dear all,
I am struggling with automatic image analysis. One of the first stumbling point seems to be segmenting DAPI-stained nuclei in fluorescence images. If this is not working reliably, the whole point of processing large numbers of images is obsolete. Attached are examples of such clumps (there are many more). If segmentation cannot separate them, it would be helpful to have at least some way of eliminating false results by just clicking on them, or even better, have all identified particles presented in a graph with freely chosen measurement as parameters as axes. Then one could click on the individual points in the graph and see the corresponding cell in the image and then decide whether to keep or discaed it. This should also work the other way round, i.e. click on a cell in the image and see the corresponding point in the graph. Similarily, it would be most helpful to be able do somethig like gating in FACS, i.e. set limits to certain parameters and see, which cells fulill them. I know that this is asking a lot, but I think providing this kind of routines in an easy to use plugin would make ImageJ even more attractive and competitive than it already is. Of course this applies only for 'Non-Cracks'. Any help would be welcome. Further questions alnosr guaranteed. Cheers P.S.: Can only insert jpeg images Matthias 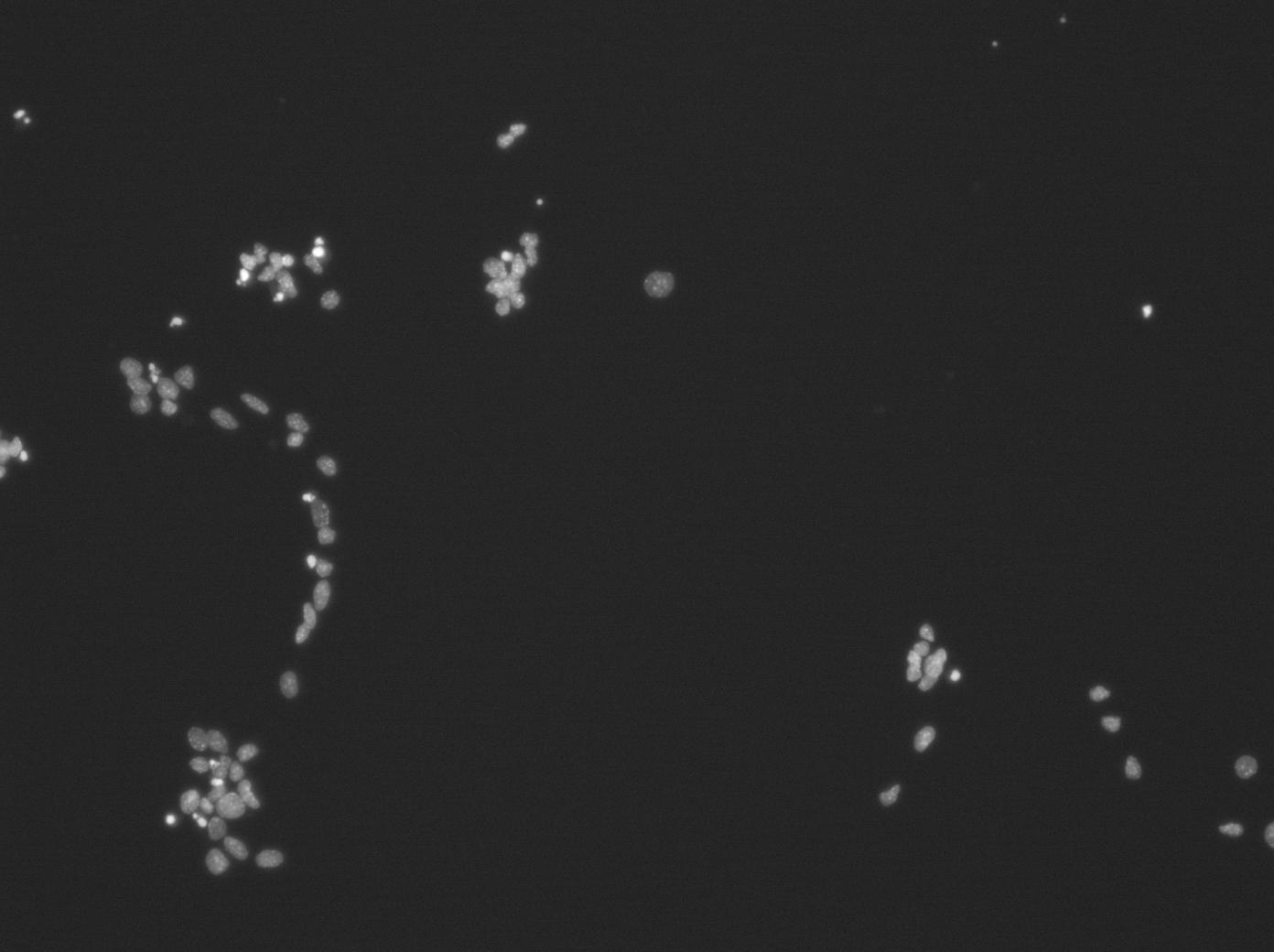
|
|
|
Dear Matthias,
Effective and reliable segmentation of the Dapi nuclei is not a trivial task when they are so clumped together. Below is script I tried for your image that identifies the easy to identify nuclei!. Basic strategy 1. background remove 2. Find maxima as first attempt to locate probable nuclei centres, then applies the result to the original image to cut apart some of the clusters 3.Auto threshold the result and perform Analyse Particles filtering out those particles that are too big (size=250-500), too small or not round enough (circularity=0.50-1.00) Matched particles are added to the ROI manager for easy access. //=================================== run("Subtract Background...", "rolling=30"); run("Find Maxima...", "noise=20 output=[Segmented Particles] exclude"); imageCalculator("Multiply create 32-bit", "RGB_(blue).jpg","RGB_(blue).jpg Segmented"); selectWindow("Result of RGB_(blue).jpg"); setAutoThreshold("Default dark"); run("Clear Results"); run("Measure"); run("Analyze Particles...", "size=250-500 circularity=0.50-1.00 show=Outlines display exclude clear add"); selectWindow("Drawing of Result of RGB_(blue).jpg"); selectWindow("Result of RGB_(blue).jpg"); //=================================== If the script is of any use, you will obviously need to modify to work with any image and not just "RGB_(blue).jpg". Hope this is of some help. -- Michael Ellis SmartCaptureLite ImageJ plugin available free now at http://www.dsuk.biz/DSUK/SmartCaptureLite.html On 21 Mar 2011, at 20:19, Matthias Kirsch wrote: > dear all, > > I am struggling with automatic image analysis. One of the first stumbling > point seems to be segmenting DAPI-stained nuclei in fluorescence images. If > this is not working reliably, the whole point of processing large numbers of > images is obsolete. Attached are examples of such clumps (there are many > more). If segmentation cannot separate them, it would be helpful to have at > least some way of eliminating false results by just clicking on them, or > even better, have all identified particles presented in a graph with freely > chosen measurement as parameters as axes. Then one could click on the > individual points in the graph and see the corresponding cell in the image > and then decide whether to keep or discaed it. This should also work the > other way round, i.e. click on a cell in the image and see the corresponding > point in the graph. Similarily, it would be most helpful to be able do > somethig like gating in FACS, i.e. set limits to certain parameters and see, > which cells fulill them. > > I know that this is asking a lot, but I think providing this kind of > routines in an easy to use plugin would make ImageJ even more attractive and > competitive than it already is. Of course this applies only for > 'Non-Cracks'. > > Any help would be welcome. Further questions alnosr guaranteed. > > Cheers > > P.S.: Can only insert jpeg images > Matthias http://imagej.588099.n2.nabble.com/file/n6193826/RGB_%28blue%29.jpg > > -- > View this message in context: http://imagej.588099.n2.nabble.com/declump-those-buggers-tp6193826p6193826.html > Sent from the ImageJ mailing list archive at Nabble.com. |
|
|
In reply to this post by Matthias Kirsch
Matthias
P.S. You might want to try out different "Analyze Particles" settings for the size and circularity options to the to find the results that best suit your needs: run("Analyze Particles...", "size=200-500 circularity=0.70-1.00 show=Outlines display exclude clear add"); --Michael Ellis On 21 Mar 2011, at 20:19, Matthias Kirsch wrote: > dear all, > > I am struggling with automatic image analysis. One of the first stumbling > point seems to be segmenting DAPI-stained nuclei in fluorescence images. If > this is not working reliably, the whole point of processing large numbers of > images is obsolete. Attached are examples of such clumps (there are many > more). If segmentation cannot separate them, it would be helpful to have at > least some way of eliminating false results by just clicking on them, or > even better, have all identified particles presented in a graph with freely > chosen measurement as parameters as axes. Then one could click on the > individual points in the graph and see the corresponding cell in the image > and then decide whether to keep or discaed it. This should also work the > other way round, i.e. click on a cell in the image and see the corresponding > point in the graph. Similarily, it would be most helpful to be able do > somethig like gating in FACS, i.e. set limits to certain parameters and see, > which cells fulill them. > > I know that this is asking a lot, but I think providing this kind of > routines in an easy to use plugin would make ImageJ even more attractive and > competitive than it already is. Of course this applies only for > 'Non-Cracks'. > > Any help would be welcome. Further questions alnosr guaranteed. > > Cheers > > P.S.: Can only insert jpeg images > Matthias http://imagej.588099.n2.nabble.com/file/n6193826/RGB_%28blue%29.jpg > > -- > View this message in context: http://imagej.588099.n2.nabble.com/declump-those-buggers-tp6193826p6193826.html > Sent from the ImageJ mailing list archive at Nabble.com. |
|
|
In reply to this post by Matthias Kirsch
Hello Matthias,
I routinely have to count cells in low magnification wide-field images and have tried many different methods to segment these images. The best one I have developed so far has an error rate below 1% for my image sets!!! Maybe it will be worth trying for your images, since they look very similar to mine. I cannot test the method on a jpg image; I need raw, uncompressed images for that. The key is using the watershed step, which separates those clumps, and a square root function to reduce differences in intensity from one nucleus to the other. No method will ever be *perfect*, of course. But counting nuclei is so tedious and boring that it would be a waste to do it by hand. If you get an error below 10% it most likely won't affect the statistical significance of your results (that depends on how many cells you are going to count and what else you need to measure). So don't give up hope for segmenting your images! Here is my method, which is normally incorporated into a BatchProcessFolders-based macro: selectImage("orig"); run("Duplicate...", "title=dup"); // I just make a copy of the image to subtract background later... selectImage("dup"); run("Gaussian Blur...", "sigma=60"); // This blurred image represents the background. imageCalculator("Subtract create", "orig", "dup"); // Subtract the background... rename("DAPIimg"); selectImage("orig"); close(); selectImage("dup"); close(); run("8-bit"); run("Gaussian Blur...", "sigma=4"); run("Remove Outliers...", "radius=10 threshold=0 which=Bright"); // Removes those dotty nucleoli run("Square Root"); // Reduces intensity differences so thresholding works better. setAutoThreshold("Triangle dark"); // The actual segmentation step. run("Convert to Mask"); run("Watershed"); // Separate the nuclei that are close together. run("Analyze Particles...", "size=914-5889 circularity=0.67-1.00 show=Nothing summarize add"); // Filter... dlength = roiManager("count"); if (dlength>0) { // If some cells were detected, save count and continue... selectWindow("Summary"); save(dest+File.separator+number+"_cellcount.txt"); // You have to modify this line for your filenames. selectWindow("Summary"); run("Close"); roiManager("reset"); run("Clear Results"); selectImage("DAPIimg"); close(); } else { roiManager("reset"); // If no cells were detected, close everything and print out the name of the image. run("Clear Results"); if (isOpen("Summary")) { selectWindow("Summary"); run("Close"); } print("Warning: image "+path+" was determined to be empty."); selectImage("DAPIimg"); close(); } Please let me know if it is not clear how to test this code on your images. Kind Regards, Elizabeth Crowell Matthias Kirsch a écrit : > dear all, > > I am struggling with automatic image analysis. One of the first stumbling > point seems to be segmenting DAPI-stained nuclei in fluorescence images. If > this is not working reliably, the whole point of processing large numbers of > images is obsolete. Attached are examples of such clumps (there are many > more). If segmentation cannot separate them, it would be helpful to have at > least some way of eliminating false results by just clicking on them, or > even better, have all identified particles presented in a graph with freely > chosen measurement as parameters as axes. Then one could click on the > individual points in the graph and see the corresponding cell in the image > and then decide whether to keep or discaed it. This should also work the > other way round, i.e. click on a cell in the image and see the corresponding > point in the graph. Similarily, it would be most helpful to be able do > somethig like gating in FACS, i.e. set limits to certain parameters and see, > which cells fulill them. > > I know that this is asking a lot, but I think providing this kind of > routines in an easy to use plugin would make ImageJ even more attractive and > competitive than it already is. Of course this applies only for > 'Non-Cracks'. > > Any help would be welcome. Further questions alnosr guaranteed. > > Cheers > > P.S.: Can only insert jpeg images > Matthias http://imagej.588099.n2.nabble.com/file/n6193826/RGB_%28blue%29.jpg > > -- > View this message in context: http://imagej.588099.n2.nabble.com/declump-those-buggers-tp6193826p6193826.html > Sent from the ImageJ mailing list archive at Nabble.com. > -- Elizabeth CROWELL ---------------------------------------------------------------------- Membrane Traffic and Cell Division Research Group Institut Pasteur 28 rue du Dr Roux 75015 PARIS, France Tel : 01.44.38.94.07 Fax : 01.45.68.89.54 ---------------------------------------------------------------------- |
|
|
> Matthias Kirsch a écrit :
> I am struggling with automatic image analysis. One of the first stumbling > point seems to be segmenting DAPI-stained nuclei in fluorescence images. > If this is not working reliably, the whole point of processing large > numbers of images is obsolete. Attached are examples of such clumps > (there are many more). If segmentation cannot separate them, it would be > helpful to have at least some way of eliminating false results by just > clicking on them, or even better, have all identified particles Without knowing exactly what you tried, saying that "it does not work" leaves a lot to be explained. There are many useful procedures that use greyscale morphology (Pierre Soille's book is an excellent source) and I would suggest looking at the book. I have implemented quite a few of the procedures described in the Morphology collection). For example, I took your image, smoothed with a gaussian filter or radius 2, then applied the procedure known as White Hat by Reconstruction with a radius of size 6. This is the macro procedure: // GreyWhiteTopHatByReconstruction // G. Landini at bham. ac. uk. // 1/Oct/2010 setBatchMode(true); a=getTitle(); run("Duplicate...", "title=_seed"); run("Minimum...", "radius=6"); run("GreyscaleReconstruct ", "mask="+a+" seed=_seed"); imageCalculator("Subtract create", a,"_seed"); selectWindow("Result of "+a); rename("WhiteTopHatReconstructed"); setBatchMode(false); For this to work you will need the Morphology plugins installed (specifically the GreyscaleReconstruct). If you threshold the result image >0 you will get lots of blobs, including those which mark the nuclei, most of these are separated. If not separated, you can apply the watershed separation to this binary image. You then threshold the smoothed original and use that binary image as a "mask" and the result of the top hat above as a "seed" to dilate the seeds without merging inside the mask. There are 2 or 3 procedures that can do this: InfluenceZones, DilateNoMerge8 or BinaryGeodesicDilateNoMerge8. The first one will not work for this example, the 2nd might do, they are both in the Morphology plugins in my site. The 3rd one (the one I used in the example) I can send it to those interested as I had no time to upload it yet. Then you AND the original and the result of the last "dilation without merging". That would preserve those dilated seeds that existed in the original thresholded image. Since the seeds were separated, the dilation without merging preserves the individual seeds. The ROIS of the result on top of the original are shown in the image attached. Not perfect, but it would narrow down the search for those clusters impossible to separate. Regards Gabriel |
|
|
In reply to this post by Matthias Kirsch
Hi Matthias
On Mar 23, 2011, at 5:00 AM, IMAGEJ automatic digest system wrote: > Date: Mon, 21 Mar 2011 13:19:39 -0700 > From: Matthias Kirsch <[hidden email]> > Subject: declump those buggers > > dear all, > > I am struggling with automatic image analysis. One of the first stumbling > point seems to be segmenting DAPI-stained nuclei in fluorescence images. If > this is not working reliably, the whole point of processing large numbers of > images is obsolete. Attached are examples of such clumps (there are many > more). If segmentation cannot separate them, it would be helpful to have at > least some way of eliminating false results by just clicking on them, or > even better, have all identified particles presented in a graph with freely > chosen measurement as parameters as axes. Then one could click on the > individual points in the graph and see the corresponding cell in the image > and then decide whether to keep or discaed it. This should also work the > other way round, i.e. click on a cell in the image and see the corresponding > point in the graph. Similarily, it would be most helpful to be able do > somethig like gating in FACS, i.e. set limits to certain parameters and see, > which cells fulill them. > > I know that this is asking a lot, but I think providing this kind of > routines in an easy to use plugin would make ImageJ even more attractive and > competitive than it already is. Of course this applies only for > 'Non-Cracks'. The reason there is not much that just works out of the box is that this is actually a very hard problem. THe one that solves it in a robust and generally applicable way will get very famous. The DNA staining is non uniform, and the nuclei are very close, so the feature you really need to get is the space between them, and in your images, that resolution simply isn't there! In amy cases even by eye, with a clever human brain , its hard to say it there are 1 or 2 nuclei in a certain blob. Clever tricks like watershed separtation only get you so far. here is a pipeline that sometimes works 0) maybe forget dapi staining.. it doesnt give you the info you actually want... where is the nucleus. find a stain that gives the spatial info you actually want. 1) Get as high resolution, blur free images as possible so you can see the gaps between the nuclei (deconvolution might help increase contrast -thats a whole story in itself) 2) find a method to smooth out the blobbyness of the nuclear staining.... without losing the edges... maybe anisotropic diffusion or some other fancy filtersing 3) compute the gradient image or some difference of gaussian or whatever feature extraction 4) use that for the object selection... fix the errors somehow. > > Any help would be welcome. Further questions alnosr guaranteed. > sorry there is nothing specific... but start by assessing if the info you want is even given by the dapi staining you are using. Its often assumed that it is, since it kinda looks like it does, but in fact it is often not the case. DAPI does not stain "the nucleus" it stains the chromatin which is heterogeneously distributed in the nucleus, and also stains the cytoplasm a little. cheers Dan > Cheers > > P.S.: Can only insert jpeg images > Matthias http://imagej.588099.n2.nabble.com/file/n6193826/RGB_%28blue%29.jpg Dr. Daniel James White BSc. (Hons.) PhD Senior Microscopist / Image Visualisation, Processing and Analysis Light Microscopy and Image Processing Facilities Max Planck Institute of Molecular Cell Biology and Genetics Pfotenhauerstrasse 108 01307 DRESDEN Germany +49 (0)15114966933 (German Mobile) +49 (0)351 210 2627 (Work phone at MPI-CBG) +49 (0)351 210 1078 (Fax MPI-CBG LMF) http://www.bioimagexd.net BioImageXD http://pacific.mpi-cbg.de Fiji - is just ImageJ (Batteries Included) http://www.chalkie.org.uk Dan's Homepages https://ifn.mpi-cbg.de Dresden Imaging Facility Network dan (at) chalkie.org.uk ( white (at) mpi-cbg.de ) |
|
|
Hi Dan, hi Matthias,
This is getting to be a very interesting thread. I'd like to make a few comments from a biologist's point-of-view. Daniel James White a écrit : > The reason there is not much that just works out of the box is that this is actually a very hard problem. > There is no point making the problem harder than it needs to be. I think the majority of ImageJ users want to use ImageJ to get data for experiments. Then the problem becomes much simpler, because you simply need a method that gives you a good estimate of the number of nuclei and that works equally well on the images from each condition you want to compare. > The DNA staining is non uniform, and the nuclei are very close, so the feature you really need to get is the space between them, > and in your images, that resolution simply isn't there! > > In amy cases even by eye, with a clever human brain , its hard to say it there are 1 or 2 nuclei in a certain blob. > I agree with that! Even by eye we cannot be sure how many nuclei there are exactly. But this is not the interesting question for the biologist. Unless your interest is developing a perfect, miraculous method to count them, all we really want to know is: Is there a difference between the control and the other experimental conditions??? If the difference between conditions is much higher than the error rate, and we can reproduce this difference in multiple experiments, then the problem is solved for all practical purposes. > here is a pipeline that sometimes works > > 0) maybe forget dapi staining.. it doesnt give you the info you actually want... where is the nucleus. > find a stain that gives the spatial info you actually want. > > 1) Get as high resolution, blur free images as possible so you can see the gaps between the nuclei (deconvolution might help increase contrast -thats a whole story in itself) > 2) find a method to smooth out the blobbyness of the nuclear staining.... without losing the edges... maybe anisotropic diffusion or some other fancy filtersing > 3) compute the gradient image or some difference of gaussian or whatever feature extraction > 4) use that for the object selection... fix the errors somehow. However, we want to use ImageJ segmentation and analysis to increase the quality of our data and to SAVE TIME... If we have to repeat the staining with a new dye, acquire perfect images at high magnification, and then run deconvolution, certainly we will spend 5 times as much time on the experiment. Then it seems faster, actually, to count the nuclei manually rather than spending so much time to optimize experimental conditions, even if the data quality is a little lower because subject to user error. So, my advice is to determine what precision and accuracy are actually needed to make a conclusion about the experiment, and then to see if some simple segmentation methods in ImageJ can give you that accuracy with the images you already have. The next time you need to repeat such an experiment, you will know how to acquire better images for a better automatic analysis. > but start by assessing if the info you want is even given by the dapi staining you are using. > Its often assumed that it is, since it kinda looks like it does, but in fact it is often not the case. > > DAPI does not stain "the nucleus" > it stains the chromatin which is heterogeneously distributed in the nucleus, and also stains the cytoplasm a little. > > Yes, that is a big disadvantage of DAPI staining. "Remove outliers" helps a little to reduce the heterogeneous staining. But what other dyes are available?? Doesn't Hoescht staining give the same result? Kind Regards, Elizabeth Crowell > cheers > > Dan > > >> Cheers >> >> P.S.: Can only insert jpeg images >> Matthias http://imagej.588099.n2.nabble.com/file/n6193826/RGB_%28blue%29.jpg >> > > Dr. Daniel James White BSc. (Hons.) PhD > Senior Microscopist / Image Visualisation, Processing and Analysis > Light Microscopy and Image Processing Facilities > Max Planck Institute of Molecular Cell Biology and Genetics > Pfotenhauerstrasse 108 > 01307 DRESDEN > Germany > > +49 (0)15114966933 (German Mobile) > +49 (0)351 210 2627 (Work phone at MPI-CBG) > +49 (0)351 210 1078 (Fax MPI-CBG LMF) > > http://www.bioimagexd.net BioImageXD > http://pacific.mpi-cbg.de Fiji - is just ImageJ (Batteries Included) > http://www.chalkie.org.uk Dan's Homepages > https://ifn.mpi-cbg.de Dresden Imaging Facility Network > dan (at) chalkie.org.uk > ( white (at) mpi-cbg.de ) > -- Elizabeth CROWELL ---------------------------------------------------------------------- Membrane Traffic and Cell Division Research Group Institut Pasteur 28 rue du Dr Roux 75015 PARIS, France Tel : 01.44.38.94.07 Fax : 01.45.68.89.54 ---------------------------------------------------------------------- |
|
|
In reply to this post by Matthias Kirsch
Dear all,
thanks a lot for all your suggestions and comments. I will work with them as soon as possible (at the moment I am packed with teaching and examination duties). I am happy to see that my post has not only stimulated some 'image analysis' discussions, but also some very basic scientifc ones. Now, to prevent misunderstandings: I do not only want to count cells. There are much easier ways to do this, even with ImageJ (I would just determine the area covered by DAPI-stained 'things' and divide that by an average nucleus area). I will attach cropped images of what I would eventually like to get at. For all the subsequent analysis steps it is mandatory to faithfully segment indivdual cells. It may even be necessary to determine the degree of clumping, as this also carries biological information. We are working with cultures of neural stem cells, which, unfortunately are somewhat heterogeneous. We fix and stain them with various antibodies after whatever treatment. The question now is: what percentage of cells shows what level of staining for a particular marker? For a start I wanted to use nuclear stains, e.g. antibodies against transcriptions factors. Some cells will have a lot, others less. E.g. it will be interesting to determine which and how many of the proliferating cells, identified by an antibody against a proliferation marker, express transcription factor x. Combining various antibodies will then help to characterize the cells with respect to treatment. Think of it like a FACS-experiment, the difference being that the cells are not in suspension (this by the way could be achieved, but would require the cells to be dissociated, which not always is a good way to go about). So for each cell in the image there should be a 'number' for its staining intensity for 'label1', 'label2', and possibly more (depending on the number of labels one can apply). The basis for all this is to faithfully identify and segment all (or most) cells in the picture. Behind all of this obviously is an attempt to do this somewhat objectively, not making it necessary to put cohorts of students at work to count all of this manually, or better put 'eye'ly. Of course it will be necessary to analyze a lot of images in batch mode, ideally unsupervised (some moderate 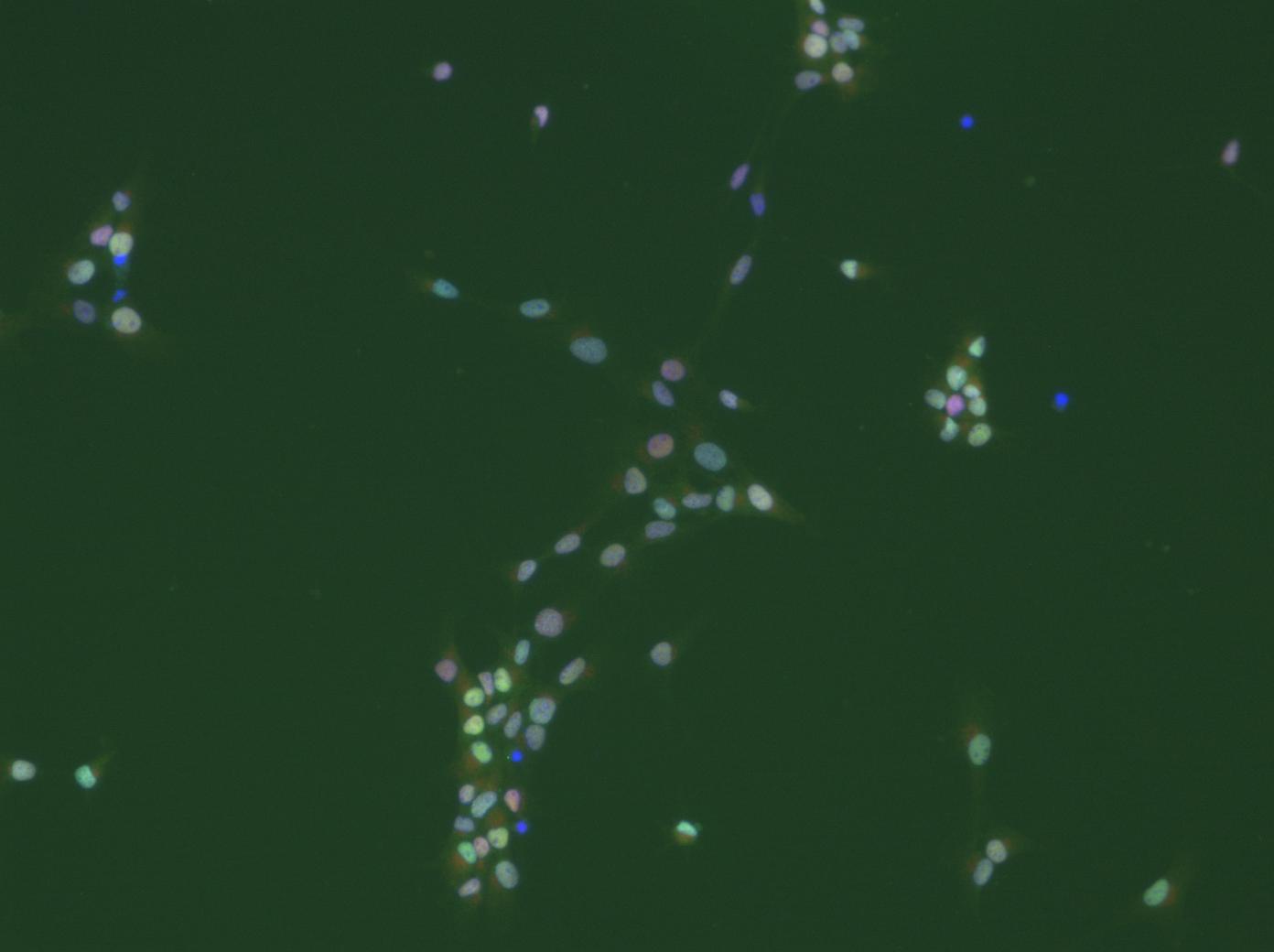 editing could be bearable at the end). If one would do a biochemical expriment, one would homogenize the cells, make a Western-Blot, normalize it to the number of cells and so on. In this way, however, information about single cells in a heterogeneous population would be lost all together. editing could be bearable at the end). If one would do a biochemical expriment, one would homogenize the cells, make a Western-Blot, normalize it to the number of cells and so on. In this way, however, information about single cells in a heterogeneous population would be lost all together.
In a next step of the analysis, we would like to include cytoplasmic markers as well, which then will make the whole business of doing all this on a single cell level even more demanding. But that's the direction, where a lot of commercial (and unaffordable) attempts are aimed at (see e.g. the Definiens software). A useful addition in this context would be an interactive way of combining graphs of combinations of measurement with the images themselves: e.g. display a scatter blot of size vs. intensity and be able to click on a point in the graph and see the corresponding cell in the image and vice versa. In addition, doing somewhat like gating in FACS would be more than helpful. All these wishes by far exceed my cababilities of constructing routines for ImageJ, which is why I am asking advice rom the vast community of 'cracks'. This much for today. I will be happy to answer more questions and will certainly wellcome any suggestions. I will attach a picture with altogether three labels (dapi in the blue channel). Quality is limited as I can only uoload jpegs. Cheers Matthias - |
|
|
In reply to this post by Matthias Kirsch
Dear all,
thanks a lot for all your suggestions and comments. I will work with them as soon as possible (at the moment I am packed with teaching and examination duties). I am happy to see that my post has not only stimulated some 'image analysis' discussions, but also some very basic scientifc ones. Now, to prevent misunderstandings: I do not only want to count cells. There are much easier ways to do this, even with ImageJ (I would just determine the area covered by DAPI-stained 'things' and divide that by an average nucleus area). I will attach cropped images of what I would eventually like to get at. For all the subsequent analysis steps it is mandatory to faithfully segment indivdual cells. It may even be necessary to determine the degree of clumping, as this also carries biological information. We are working with cultures of neural stem cells, which, unfortunately are somewhat heterogeneous. We fix and stain them with various antibodies after whatever treatment. The question now is: what percentage of cells shows what level of staining for a particular marker? For a start I wanted to use nuclear stains, e.g. antibodies against transcriptions factors. Some cells will have a lot, others less. E.g. it will be interesting to determine which and how many of the proliferating cells, identified by an antibody against a proliferation marker, express transcription factor x. Combining various antibodies will then help to characterize the cells with respect to treatment. Think of it like a FACS-experiment, the difference being that the cells are not in suspension (this by the way could be achieved, but would require the cells to be dissociated, which not always is a good way to go about). So for each cell in the image there should be a 'number' for its staining intensity for 'label1', 'label2', and possibly more (depending on the number of labels one can apply). The basis for all this is to faithfully identify and segment all (or most) cells in the picture. Behind all of this obviously is an attempt to do this somewhat objectively, not making it necessary to put cohorts of students at work to count all of this manually, or better put 'eye'ly. Of course it will be necessary to analyze a lot of images in batch mode, ideally unsupervised (some moderate 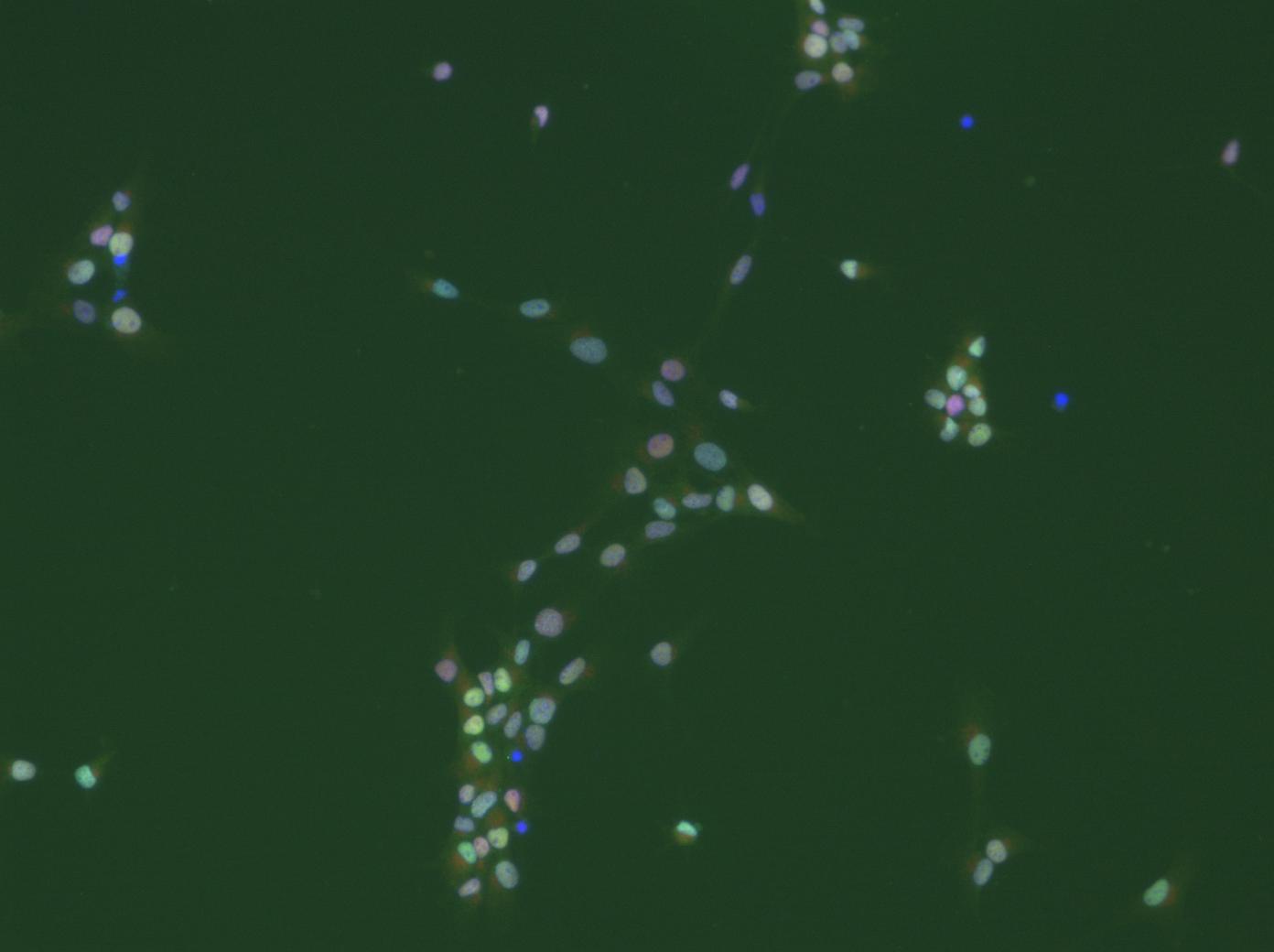 editing could be bearable at the end). If one would do a biochemical expriment, one would homogenize the cells, make a Western-Blot, normalize it to the number of cells and so on. In this way, however, information about single cells in a heterogeneous population would be lost all together. editing could be bearable at the end). If one would do a biochemical expriment, one would homogenize the cells, make a Western-Blot, normalize it to the number of cells and so on. In this way, however, information about single cells in a heterogeneous population would be lost all together.
In a next step of the analysis, we would like to include cytoplasmic markers as well, which then will make the whole business of doing all this on a single cell level even more demanding. But that's the direction, where a lot of commercial (and unaffordable) attempts are aimed at (see e.g. the Definiens software). A useful addition in this context would be an interactive way of combining graphs of combinations of measurement with the images themselves: e.g. display a scatter blot of size vs. intensity and be able to click on a point in the graph and see the corresponding cell in the image and vice versa. In addition, doing somewhat like gating in FACS would be more than helpful. All these wishes by far exceed my cababilities of constructing routines for ImageJ, which is why I am asking advice rom the vast community of 'cracks'. This much for today. I will be happy to answer more questions and will certainly wellcome any suggestions. I will attach a picture with altogether three labels (dapi in the blue channel). Quality is limited as I can only uoload jpegs. Cheers Matthias - |
|
|
In reply to this post by Gabriel Landini
Dear all,
I thought lets have a go at this as well. I did the following: run("Rolling Ball Background", "rolling=30"); setAutoThreshold("Default dark"); setThreshold(42, 255); run("Convert to Mask"); run("Close"); run("Watershed"); What gave finally the attached result RGB_(blue)-2.jpg You might be able to improve this by dupplicating your image after the Rolling Ball Background and continue with the duplicated image: run("Find Maxima...", "noise=30 output=[Segmented Particles]"); imageCalculator("Add create 32-bit", "RGB_(blue)-1.jpg","RGB_(blue).jpg Segmented"); setAutoThreshold("Default"); run("Convert to Mask"); setThreshold(255, 255); run("Convert to Mask"); run("Analyze Particles...", "size=15-Infinity circularity=0.00-1.00 show=Outlines add"); This will result in image RGB_(blue)-3.jpg Still not exactly correct...... Good luck Kees ________________________________________ From: ImageJ Interest Group [[hidden email]] On Behalf Of Gabriel Landini [[hidden email]] Sent: 23 March 2011 09:44 To: [hidden email] Subject: Re: declump those buggers > Matthias Kirsch a écrit : > I am struggling with automatic image analysis. One of the first stumbling > point seems to be segmenting DAPI-stained nuclei in fluorescence images. > If this is not working reliably, the whole point of processing large > numbers of images is obsolete. Attached are examples of such clumps > (there are many more). If segmentation cannot separate them, it would be > helpful to have at least some way of eliminating false results by just > clicking on them, or even better, have all identified particles Without knowing exactly what you tried, saying that "it does not work" leaves a lot to be explained. There are many useful procedures that use greyscale morphology (Pierre Soille's book is an excellent source) and I would suggest looking at the book. I have implemented quite a few of the procedures described in the Morphology collection). For example, I took your image, smoothed with a gaussian filter or radius 2, then applied the procedure known as White Hat by Reconstruction with a radius of size 6. This is the macro procedure: // GreyWhiteTopHatByReconstruction // G. Landini at bham. ac. uk. // 1/Oct/2010 setBatchMode(true); a=getTitle(); run("Duplicate...", "title=_seed"); run("Minimum...", "radius=6"); run("GreyscaleReconstruct ", "mask="+a+" seed=_seed"); imageCalculator("Subtract create", a,"_seed"); selectWindow("Result of "+a); rename("WhiteTopHatReconstructed"); setBatchMode(false); For this to work you will need the Morphology plugins installed (specifically the GreyscaleReconstruct). If you threshold the result image >0 you will get lots of blobs, including those which mark the nuclei, most of these are separated. If not separated, you can apply the watershed separation to this binary image. You then threshold the smoothed original and use that binary image as a "mask" and the result of the top hat above as a "seed" to dilate the seeds without merging inside the mask. There are 2 or 3 procedures that can do this: InfluenceZones, DilateNoMerge8 or BinaryGeodesicDilateNoMerge8. The first one will not work for this example, the 2nd might do, they are both in the Morphology plugins in my site. The 3rd one (the one I used in the example) I can send it to those interested as I had no time to upload it yet. Then you AND the original and the result of the last "dilation without merging". That would preserve those dilated seeds that existed in the original thresholded image. Since the seeds were separated, the dilation without merging preserves the individual seeds. The ROIS of the result on top of the original are shown in the image attached. Not perfect, but it would narrow down the search for those clusters impossible to separate. Regards Gabriel |
|
|
In reply to this post by Matthias Kirsch
Hi Matthias,
have you ever thought about using the RGB color information as an additional input for the cell separation? It looks quit promising. Peter Am 23.03.2011 20:04, schrieb Matthias Kirsch: > Dear all, > > thanks a lot for all your suggestions and comments. I will work with them as > soon as possible (at the moment I am packed with teaching and examination > duties). > > I am happy to see that my post has not only stimulated some 'image analysis' > discussions, but also some very basic scientifc ones. Now, to prevent > misunderstandings: I do not only want to count cells. There are much easier > ways to do this, even with ImageJ (I would just determine the area covered > by DAPI-stained 'things' and divide that by an average nucleus area). I will > attach cropped images of what I would eventually like to get at. For all the > subsequent analysis steps it is mandatory to faithfully segment indivdual > cells. It may even be necessary to determine the degree of clumping, as this > also carries biological information. > We are working with cultures of neural stem cells, which, unfortunately are > somewhat heterogeneous. We fix and stain them with various antibodies after > whatever treatment. The question now is: what percentage of cells shows what > level of staining for a particular marker? For a start I wanted to use > nuclear stains, e.g. antibodies against transcriptions factors. Some cells > will have a lot, others less. E.g. it will be interesting to determine which > and how many of the proliferating cells, identified by an antibody against a > proliferation marker, express transcription factor x. Combining various > antibodies will then help to characterize the cells with respect to > treatment. Think of it like a FACS-experiment, the difference being that the > cells are not in suspension (this by the way could be achieved, but would > require the cells to be dissociated, which not always is a good way to go > about). So for each cell in the image there should be a 'number' for its > staining intensity for 'label1', 'label2', and possibly more (depending on > the number of labels one can apply). The basis for all this is to faithfully > identify and segment all (or most) cells in the picture. Behind all of this > obviously is an attempt to do this somewhat objectively, not making it > necessary to put cohorts of students at work to count all of this manually, > or better put 'eye'ly. Of course it will be necessary to analyze a lot of > images in batch mode, ideally unsupervised (some moderate > http://imagej.588099.n2.nabble.com/file/n6201542/RGB.jpg editing could be > bearable at the end). If one would do a biochemical expriment, one would > homogenize the cells, make a Western-Blot, normalize it to the number of > cells and so on. In this way, however, information about single cells in a > heterogeneous population would be lost all together. > In a next step of the analysis, we would like to include cytoplasmic markers > as well, which then will make the whole business of doing all this on a > single cell level even more demanding. But that's the direction, where a lot > of commercial (and unaffordable) attempts are aimed at (see e.g. the > Definiens software). > A useful addition in this context would be an interactive way of combining > graphs of combinations of measurement with the images themselves: e.g. > display a scatter blot of size vs. intensity and be able to click on a point > in the graph and see the corresponding cell in the image and vice versa. In > addition, doing somewhat like gating in FACS would be more than helpful. > All these wishes by far exceed my cababilities of constructing routines for > ImageJ, which is why I am asking advice rom the vast community of 'cracks'. > > This much for today. I will be happy to answer more questions and will > certainly wellcome any suggestions. I will attach a picture with altogether > three labels (dapi in the blue channel). Quality is limited as I can only > uoload jpegs. > > Cheers > Matthias > - > > -- > View this message in context: http://imagej.588099.n2.nabble.com/declump-those-buggers-tp6193826p6201542.html > Sent from the ImageJ mailing list archive at Nabble.com. > |
|
|
In reply to this post by Matthias Kirsch
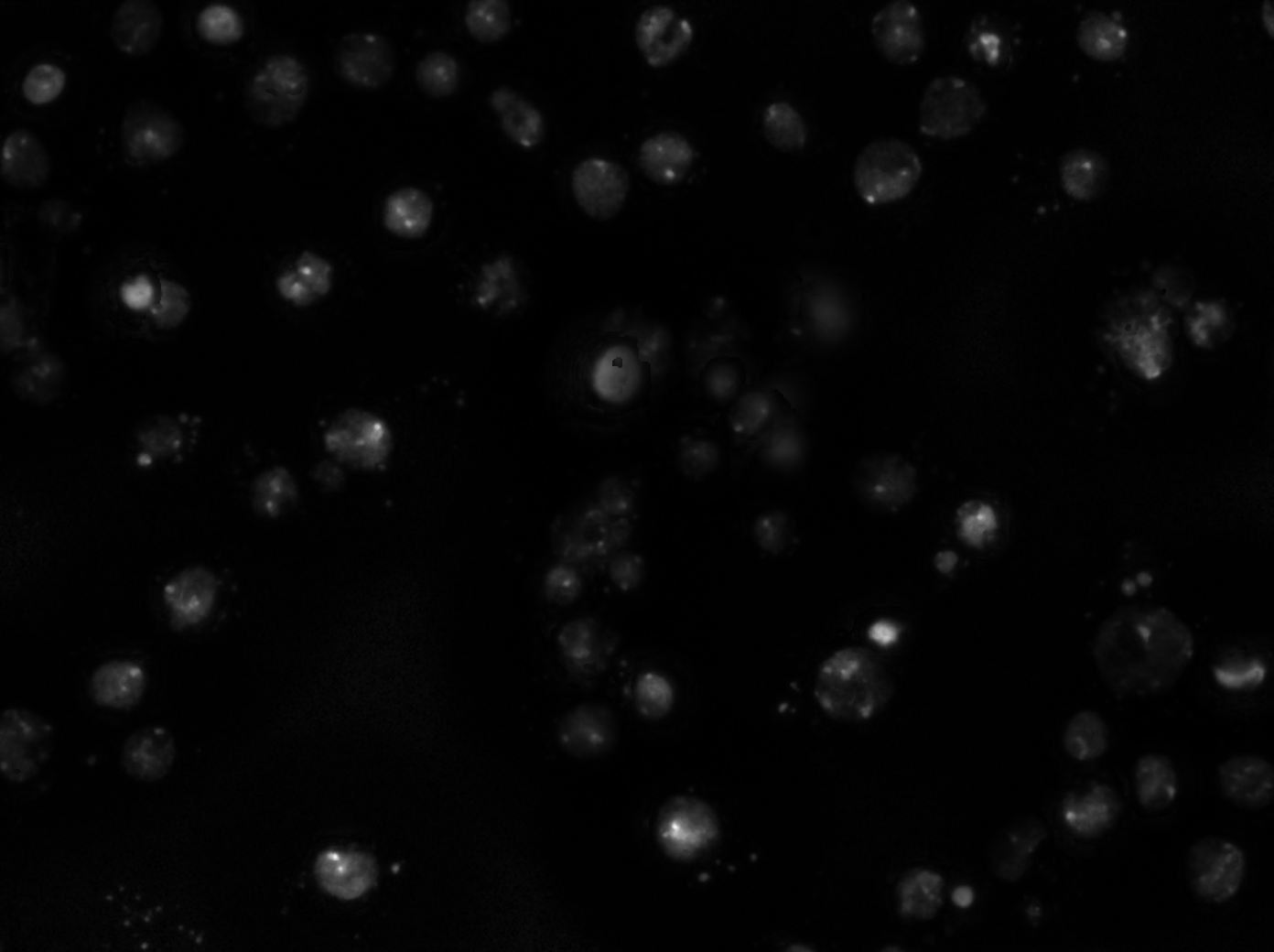 Any suggestions for my particular case? Currently, I am only the programmer, so I am unable to change the stains used, type of cells, etc. I personally think that the Watershed program is inherently flawed. I've been working on a new version. I was trying to stick to high level commands offered by imageJ to create the new version, but I am thinking that I will actually have write a plugin eventually. |
«
Return to ImageJ
|
1 view|%1 views
| Free forum by Nabble | Edit this page |